第2部 徹底討論
「薬の承認制度を知ろう-薬機法改正の問題点と課題-」概要報告
特定非営利活動法人ネットワーク医療と人権 事務局 景山 千愛
(前頁からの続き)
●医薬品の有効性、有用性
医薬品が承認されるためには、医療等で使えなければなりません。ここでいう「使える」という意味は一義的には「効能効果がある」ということです。しかしながら、ある解熱剤があって「熱は1 秒で下がるけれど、そのあと消化管に穴が開きます」ということでは、「使える」とは言えません。したがって、医薬品として承認されるためには、効能効果があるだけでは不十分で、効能効果が害作用(= 副作用)を上回っている事が必要になります。ここで、初めて有効性が確認されたことになるわけです。製薬企業が医薬品として売りたいと言い出さない限り(承認申請をしない限り)、世の中に医薬品が増えることはないのです。
加えて「使える」という意味には、有用性という問題も含まれます。例えば「1 秒で熱が下がり、しかも副作用が少ない解熱剤があっても、1 錠30 万円」となったら使えない(=有用性がない)のです。
有用性の議論で、HTA(Health Technology Assessment)という言葉が世界的に流通していて、医療技術をどのように使うのかという、使い方の議論があります。有用性と有効性は厳密には区別されます。
●医薬品の処方と医療費
日本の場合は、医薬品が承認されれば、ほぼ保険収載されるので、例えば、がんに関する知識があまりない医師であっても、保険者が保険償還の条件を付さない限り、すべての抗がん剤を処方できます。
日本の国民皆保険制度では、医療機関が行った医療行為等に診療報酬として点数がついていて、事後に保険者から医療機関にお金が支払われます。この健康保険でカバーするかどうかが、医療政策上非常に重要な位置を占めていて、医薬品を適切に使用するための議論をする場合、有用性の観点を無視する事はできません。
抗がん剤のオプジーボや、C 型肝炎ウイルスの抗ウイルス薬ハーボニーはかなり高額な医薬品です。日本では、医療における費用対効果の観点から、健康保険でこれらの薬を使える医療機関や対象となる患者を限定する例が最近見られるようになりました。
●臨床試験の流れとメーカーの思惑
医薬品が使えるか使えないかを国に諮るのに、メーカーに何が必要かというと、その医薬品としての有効性を示す必要があります。統計的に有効性があるということを証明するデータです。
そのデータを用意するプロセスが、ヒトを対象にした臨床試験=治験です。治験の前段階として効能物質を探索する基礎研究があります。研究成果としていいシーズ(化学物質)を見つけて、「これ薬になるんじゃない」となった時に、まず動物を使って毒性とか基本的な薬効を調べます。ネズミが10 匹中5 匹死にましたとか、このくらいの量だと危ないとか、もう少し量を減らさないといけないとか、人間の体重だとこのくらいかなとか探りをつけるわけです。
健康な人を対象にして、動物実験のデータを元にヒトへの作用を調べるのが第1相試験です。これをだいたい3 年から5 年くらいかけて実施します。次の第2 相試験では、少数の患者さんを対象に量を変えてみたり期間を変えてみたりして、効果を見ながら適正な量かどうかを調べます。その後の第3 相試験では、より多くの患者さんに試します。第3 相試験ではランダム化二重盲検試験と言って、ランダムに選んだ対象者を2 つのグループに分けて、片方のグループには偽薬(プラセボ)を、もう片方には試験薬を、患者さんも医者もわからないように投与して、統計学的に有効性があるか調べるのです。
「効能効果が副作用を上回って、薬としての価値がある」というエビデンスのある臨床試験データをそろえて、日本ではPMDA(医薬品医療機器総合機構)に承認申請を出します。これらの期間を合わせるとだいたい10 年から15 年、大急ぎでやっても10 年はかかります。200 億とか300 億とか、かなりの予算が必要で、メーカーの負担が大きく大変です。逆に言えば、メーカーとしては大変な手間・労力・経費を投じたのだから、なんとかして医薬品として認可してもらい、投資額を回収したいという強い動機づけがあります。多額の利益を産まないとやっていけないという事情は注意すべきです。
●薬機法改正に向けた議論の内容
薬機法改正に向けた議論は、厚生科学審議会・医薬品医療機器制度部会で行われています。
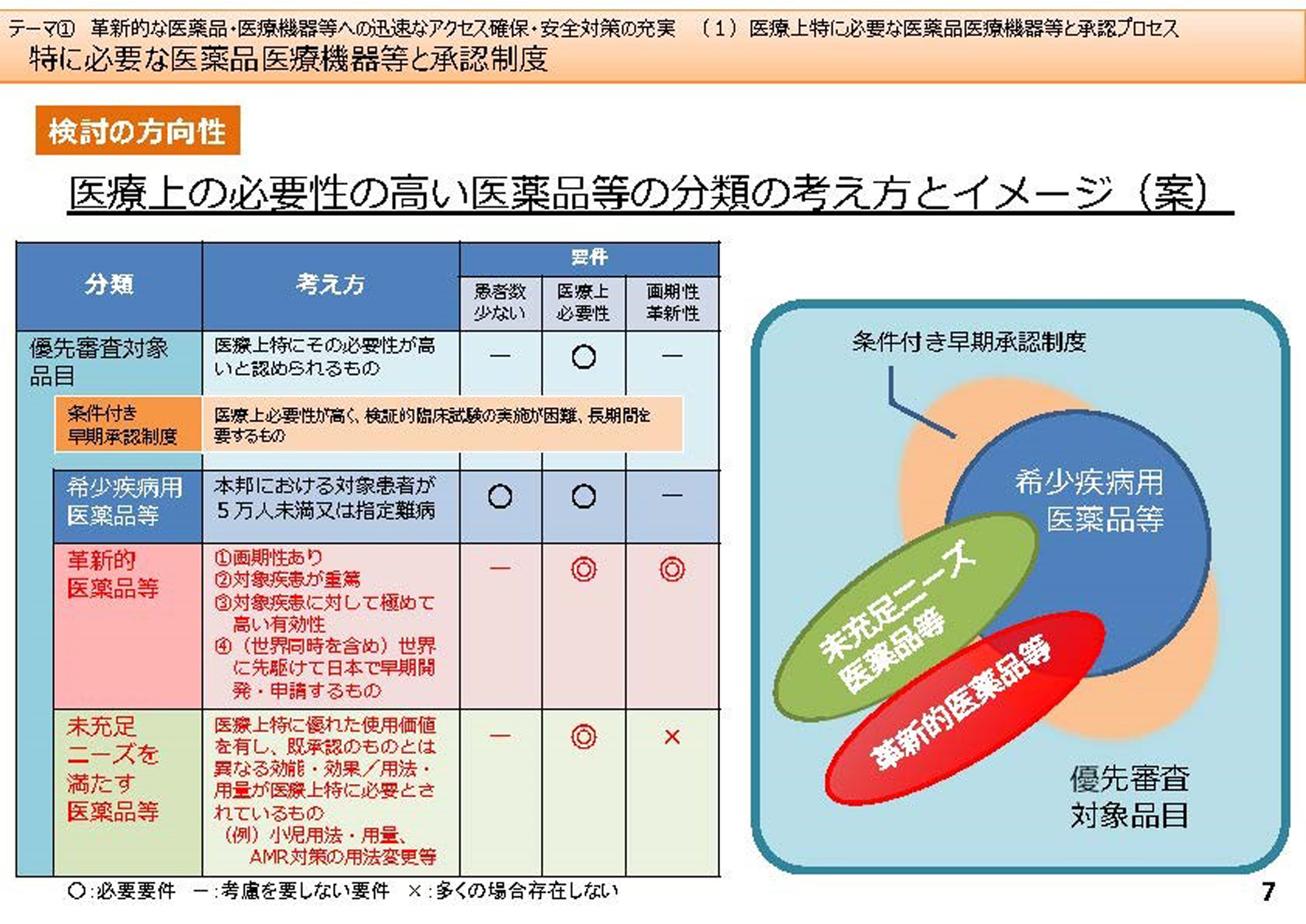
今回の改正に向けた議論の内容の1 つは、「先駆け指定」で、日本で最初に開発された画期的な新薬を優遇しようという制度です。今までは優先審査品目として1990年代に設定した希少疾病用医薬品だけが位置付けられていました。その他にも、医療上必要な医薬品とか、抗HIV 薬など、承認条件をいろいろ付けて優先審査を行い、国内では治験を行わずに承認していました。
2 つ目の議論の内容は、Unmet Medical Needs(いまだに治療法が見つかっていない疾患に対する医療ニーズ)や小児を対象とする医薬品などに対しても、医薬品の開発を進めなければならないということです。
「その他」の議論では、その部分を整理することになりました。現状は優先審査項目という全体があって、その中に患者数5 万人未満のいわゆるオーファンドラッグ(希少疾病用医薬品)が優先品目として位置づけられています。5 万人以下といっても、例えば日本では、血友病患者は5-6000 人、HIV 患者は2 万6000 人くらいですから、これらの治療薬はオーファンドラッグになります。
●承認条件と評価
何もかも早期承認していいという話にはならないのです。例えば医療上の必要性が高いか、患者が少ないか、革新性があるか、という3 つの条件があります。
例えばオーファンの場合は患者が少ないことと、治療薬の必要性で、画期性は特に問いません。革新的医薬品に関しては、患者さんが少ないことは問わず、むしろ医療上の必要性と画期性が条件になります。
それからUnmet Medical Needs に関しては、医療上の必要性があることとなっていて、条件付き早期承認制度での対応を考えています。
【図3】厚生科学審議会 平成30年度第7回医薬品医療機器制度部会 資料1
「探索的」というのが第1 相、第2 相試験で、第3 相試験後に承認申請を行なうのが通常の医薬品です。それを今回の改正では、第1 相、第2 相でいいという議論になっています。
患者が少なくて1000 人とか、再生医療などでも患者は10 人程度ですから、治療で腫瘍が小さく(少なく)なったとか、10 人中6 人は相当良好だとか、有効性が「ありそうだな」という結果= 奏功率をもって、医薬品として承認して、承認条件として、市販後の有効性・安全性確認や使用上の要件を求めるというものです。また安全性に関する中間的な評価も検討されています。
●条文化の議論について
平成29 年10 月20 日の段階の通知では、対象品目は以下に該当する医薬品と書かれています。
(1) 生命に重大な影響のある疾患、病気の進行が不可逆的で著しい影響を及ぼす疾患、その他
(2) 既存の治療法、予防法、診断法がないこと
(3) 検証的臨床試験の実施が困難、または患者数が少ないこと、実施に相当の期間を要する
(4) 検証的臨床試験以外の臨床試験等の成績から有効性、安全性が示される
しかしながら、このような内容では、法律の条文としてはあいまいなので、このまま改正を進めてよいのかという議論があります。
パネリストからの発言・問題提起
各パネリストからは、それぞれの立場で問題提起がなされました。以下は、発言の概要です。
<増山氏:制度をめぐる理想と現実の乖離>
増山氏は、サリドマイド被害当事者、一般用医薬品販売の審議会などに関わってこられました。この経験から、医薬品という商品の独自性や、制度の改正は必ずしも理想の実現に結びつかないといった問題の指摘がなされました。
(1)医薬品は、家電製品といった一般の商品とは異なる側面を持ちます。まず、ユーザーである患者が商品を「評価」することが難しいことにあります。医薬品の開発は産(民間企業)、承認は官(国・厚生労働省)、医薬品のユーザーは患者(消費者)です。しかし消費者として薬の「評価」が可能かというと非常に難しいです。さらに、医薬
品は、患者の犠牲を前提として成り立ってきた商品でもあります。
(2)これまでの薬害事件を振り返ると、ほとんどの薬害事件では、発売後に見つかった未知の重篤な副作用が放置されることで、たくさんの被害者を出しました。これまでの薬害事件を振り返ると、スモンとキノホルムの関係を否定する人は今はもうほとんどいませんし、サリドマイドと胎児の催奇形性に因果関係がないという人はいません。しかしサリドマイドは裁判が11 年間続きましたが、その半分は、原告が勝つのが難しいと言われるほど、誰も原告の立場で証言してくれる人はいませんでした。
(3)2000 年以降、一般用販売医薬品制度は何度も見直されてきました。そのなかで、医薬品販売の現実は、当時の理想とかけ離れてきています。コンビニでの医薬品販売など、さまざまな規制緩和が段階的に盛り込まれました。しかし、結局は、医薬品を売りやすく、服用してもらいやすい環境の整備につながっていくだけでした。改正時に描いていた理想が、実際に具体化すると違うものになってしまいます。
以前の薬事法改正では、薬剤師や登録販売者が購入者と対面販売を行うので安全であると言われていました。しかし現実には、この制度も非常に使い勝手の悪いものになっています。例えば、登録販売者は業務に忙殺されていること、相談スペースを持つ店舗がほとんどないことなどがあります。
陣痛促進剤も、最初はお産をコントロールするために開発されたわけではなかったかもしれません。しかし実際には現在そのような使われ方をしています。このような例を見ても、単に医薬品や薬機法、医療行政や製薬会社の実験・治験のプロセスなどを個別にとらえるのでは不十分です。制度を見直す場合、それぞれが絡みあって安全を確保していくことを、考慮に入れる必要があります。
<泉氏:有効性の再検証と罰則規定>
泉氏は、ご家族を薬害C 型肝炎で亡くされたことから、薬害C 型肝炎事件の検証や、再発防止のための活動を進めてこられました。この経緯から、副作用報告義務や安全管理に関する罰則規定などの制度が活用されていないことへの問題提起がなされました。
(1)早期承認制度はそれなりに必要です。しかし、その制度を監視・管理するための仕組みが不十分です。早期承認された後の有効性の追跡をどのように確認するのでしょうか。あとで有効性を評価するまでに、被害が出たときにどうするのでしょうか。
(2) 副作用報告義務やリスク管理計画も不十分です。せっかく救済制度が作られても、そこに救済できるだけの情報が集まっているのでしょうか。副作用報告をしなければならない医師や薬剤師が本当に報告制度を知っているのか。製薬企業によって作成されたリスク管理計画も、現実には活用されていません。リスク管理計画を活用している数値があまりにも低いということで、これはなんとか解決していかなければならないのです。
(3)医薬品の製造、安全管理に関する罰則規定も、厳格に機能しているとはいいがたいです。例えば、この「製剤・医薬品は単一企業による供給(販売)で、しかも絶対必要な薬だから」といった理由で、医薬品の供給停止は避けられてきました。いままで、製造や安全管理に違反した製薬企業も、業務停止になることはありませんでした。
(4) 日本はいま専門医の先生が非常に少ないことも問題です。専門医制度がないので、医師であればどの薬も出せます。この危うさを考えてもらいたいです。
<高町氏:利益優先主義と法の抜け穴>
高町氏は、キノホルム服用によるスモン被害という体験を通して、医薬品に関する法律や制度には必ず抜け穴があり、製薬企業は利益優先のなかで抜け穴をかいくぐるものであると指摘しました。
(1)スモンの原因となったキノホルムは、もともと劇薬指定されており、慎重な投与が行われていました。しかし第二次世界大戦のなかで、兵士を少しでも早く治療して戦場に戻したいということで、この劇薬指定が外されていき、戦後の日本では、安くてよく効く薬として売られることとなりました。つまり、有効性があって、リスクもあると分かっていて、そのリスクを回避するための手立てがなされていたとしても、何らかの理由で指定が簡単に外されていったのです。医薬品をめぐる制度には、そのような危なさ、もろさがあります。
(2)臨床試験には莫大な費用がかかり、企業にとっては負担です。第3 相試験をやめることでコストを下げて、早く承認すればベネフィットは上がります。こういった状況が、ドラッグ・ラグを解消するという口実の下にできてしまうのではないでしょうか。早期承認の裏に、利益優先が隠れているのではないかという危惧を持っています。
(3)スモンが起こったことで、1979 年の薬事法改正で初めて国の権限を強化しました。薬事法は理論上は救済法になるかもしれないですが、穴がたくさんありました。1979 年からもう40 年も経っているなかで、薬害の構造がほとんど変わっていないのは、やはり、その時には考えられなかったような抜け穴がたくさんあるためです。利益相反とか、企業が製品を売るために戦略を考えていく中で、いろいろと抜け穴を見つけてしまって、それを解決しきれていません。
司会・花井氏
確かに、「第3 相試験を省略していいなんてことは言っていません」と、国も言っています。「本当に必要な医薬品を省略するだけですよ」というのが今の制度の建付けで、制度の目的についてはみんな総論賛成だと思います。
ところがいま高町さんが指摘したのは、そのように改正しても、一旦改正してしまうと穴が開きかねないということです。それについて懸念する部分が1 つあります。いま流行りの言葉でリアルワールド・データという言葉があります。臨床試験1000人といっても、年齢層も限られていて合併症もない、要するに「きれいな」患者さんで第3 相試験をするが、実際に市販されると、がんに罹患していたり、血圧が高かったりとか、糖尿病気味、高齢、いろんな状況の人がいるので、条件が全然違うのだから、実臨床のデータも応用して調べるという話です。
しかしリアルワールド・データが真実を表しているという話がどんどん広がれば、「結局、第3 相試験をやっても無駄」とメーカーは言い出しかねません。
質疑応答
増山氏
花井さんは今、この制度についてどう思っているのでしょうか。やはりHIV 治療の中ではかなり近いところにいると思います。
司会・花井氏
HIV 医療では条件付き早期承認という形で、治験をほとんど行わずに薬を承認しています。また、HIV 医療における早期承認は、非常にうまくいっているが、それはHIV 専門の医者と患者会のコミュニティが小さく、専門的な領域に限定しているということの結果論です。
結論として早期承認という制度は必要だと思います。問題は、それにどのような条件をつけるかということです。現在の通知文では、条件があまりにも甘いので、きわめて限定的な形で法的に運用することを前提にすれば、制度化については必ずしも反対ではありません。
増山氏
ドラッグ・ラグも、PMDA のデータに示されているものによれば、もちろん病気の種類によっては若干あるけれども、ほとんど解消されていると理解しています。ただ、申請ラグはあり、本来必要なものだけれども開発してもペイしないために置き去りにされているところはあると思います。
しかし今までのパターンだと、「そういうことが解消できるのです」と言ってふたを開けてみると、やはり企業がやりたいものをやるというのが、これまで繰り返されてきたことです。日本が早期承認で新しいタイプの薬の開発を積極的にできるほどの体力があるのかも疑問です。
司会・花井氏
結局、開発ラグというのは企業がやりたいかやりたくないかの話になります。日本国内ではやりたくないという流れがあるのに、薬機法改正の「先駆け審査指定制度」で優遇して企業にやる気を出させるという話です。
臨床研究を行なう環境、大学での治験環境などをきちんとしないで、付け焼刃で「じゃあ早く申請してください」でいいのかというのは根本的テーマです。
参加者
(1)HPV ワクチンが承認されたのは、この早期承認制度を使ったものでした。めったにならないがんであるために希少疾患に入ってしまい、スピード認可されて安全確認されずに世界中で問題が起こりました。ただ、それを利用してコマーシャルなどで、アメリカとニュージーランドだけが世界の中でDirect to Consumer Advertisemen(t 処方箋を介さずに、直接消費者が医薬品を購入できる制度)が言い出されているので確認が必要です。
問題なのは、HPV ワクチンはパップテスト(子宮頸がんを発見するために使われる細胞診検査)を一緒にやらないと意味がないと言って出てきたのに、パップテストは日本では全然保険ではカバーされていないことです。
(2)日本で一番足りないと思うのは、コストパフォーマンスの分析がないということと、先進国の中で唯一医者の更新制度がないことです。知り合いの薬剤師で、医師のめちゃくちゃな処方を見てうつになりそうな人たちがたくさんいます。困ったことに薬剤師は会社員なので、目の前にある大きな病院のおかげで生きていけるのです。おかしい、殺人的だと思っても、それを問題視してしまったら、「じゃあ隣の薬局に行ってもらう」となってしまって、会社から怒られることになります。アメリカのように、薬剤師が力を持つ状態を、制度的に採り入れなければなりません。
基本的に医者が悪いとか薬剤師が悪いのではなくて、そもそもシステムがそうさせていると考えたほうがいいと思います。
(3)結局被害に遭った人だけの問題にしてしまうと、国民の声が聞けません。「あなたたちの税金が、こんなふうに無駄に使われていますよ」、「あなたたちの将来の介護費用がなくなりますよ」と、みんなの問題にしていく訴えが必要だと思います。
司会・花井氏
(1)ワクチンが「予防薬」と書いてあったので、救済制度でカバーすることはまかりならない、という件は、厚労省は了解していると主張しています。ワクチンは公衆衛生行政で厚生労働省「健康局」が緊急だと言えば承認せざるをえないので、「医薬・生活衛生局」は関係ありません。大臣がこれは必要だと承認しなければ仕方がないので、ここでわざわざ緩める必要はないという考え方です。
(2)医政制度についてはおっしゃる通りで、点数が何点だったら免許を飛ばしてしまうという制度を補充すれば、簡単に解決することが山ほどあります。しかし日本の医政統制、つまり医政局の医師法が、強い圧力団体がいて触れられないため、結果的に薬機法と健康保険法が肥大化しているという、いびつな形になっています。
(3)医政行政が空洞化しているので、全体をシステムとして構想しがたいです。だからそれを国民の意見として言うためには、やはり医療問題が政治家の票にならないといけません。ロンドン五輪のときに皆保険制度が国民の誇りとしてアピールされていました。日本はこれだけの皆保険制度があるけど、それを東京オリンピックは私たちの誇りですという感じで出す感覚があるかといえば、たぶんないでしょう。市民社会の感覚のなかに医療が薄いというのもあります。
薬害根絶フォーラムに参加した所感
薬害根絶フォーラムには初参加でしたが、席に空きがないほど会場がいっぱいだったのが印象的でした。
多くの薬害は知識として知っていたつもりでした。しかし実際に被害に遭った方々の報告を聴くと、文字情報では得られない、それぞれの薬害事件の重みを感じました。また、「薬害を繰り返さない」という目的を実現するためには、実際的で複雑な問題に直面せざるを得ないのだと実感しました。討論にあったように、より安全な医薬品を求める手続きは、医薬品のコストや承認の速度の問題と切り離せません。さらに、行政の制度や業界の利害だけでなく、ほかの患者の治療にもかかわる可能性があります。「よい・悪い」など単純に割り切って結論を出せる問題ではないため、(大変ですが)継続的に監視し、声を届けるシステムが必要であると感じました。「徹底討論」の意義もそこにあると思いました。