イントロダクション
2022年5月10日の参議院厚生労働委員会にて、MARS理事である花井十伍が参考人として招致され、薬機法一部改正について発言しました。発言概要は下記のとおりです。
参議院 厚生労働委員会(第十二回) 参考人発言より(一部補足あり)
医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律等の一部を改正する法律案(閣法第四二号)(衆議院送付)
開会日:2022年5月10日
参考人:花井十伍
(特定非営利活動法人ネットワーク医療と人権 理事/全国薬害被害者団体連絡協議会 代表世話人)
以下、参考人発言
本日はこのような機会を与えていただきありがとうございます。私は特定非営利活動法人ネットワーク医療と人権の理事の花井十伍と言います。このNPOは薬害HIV訴訟大阪原告団の呼びかけによって作られたNPO法人です。主に薬害や人権問題に関する調査研究事業、薬害エイズ被害者の支援、そして患者会の支援などを行なっているNPOです。
また全国薬害被害者団体連絡協議会(薬被連)は、10薬害12団体によって構成されています。本日のお話は、残念ながら、これら12団体の意志統一が時間的に難しいので概ね薬害エイズ被害者あるいは患者としての立場から個人的な見解だと受け止めていただければと思います。
薬害という概念自体は日本固有の概念です。英訳するとDrug induced sufferingsとなります。例えば薬害エイズ被害者は世界的にいて交流していますが、サリドマイド被害者とは交流していません。日本の場合は薬害概念と、それぞれの被害経験世界に共感があり、このような12団体が薬害根絶を目指して活動を展開しております。
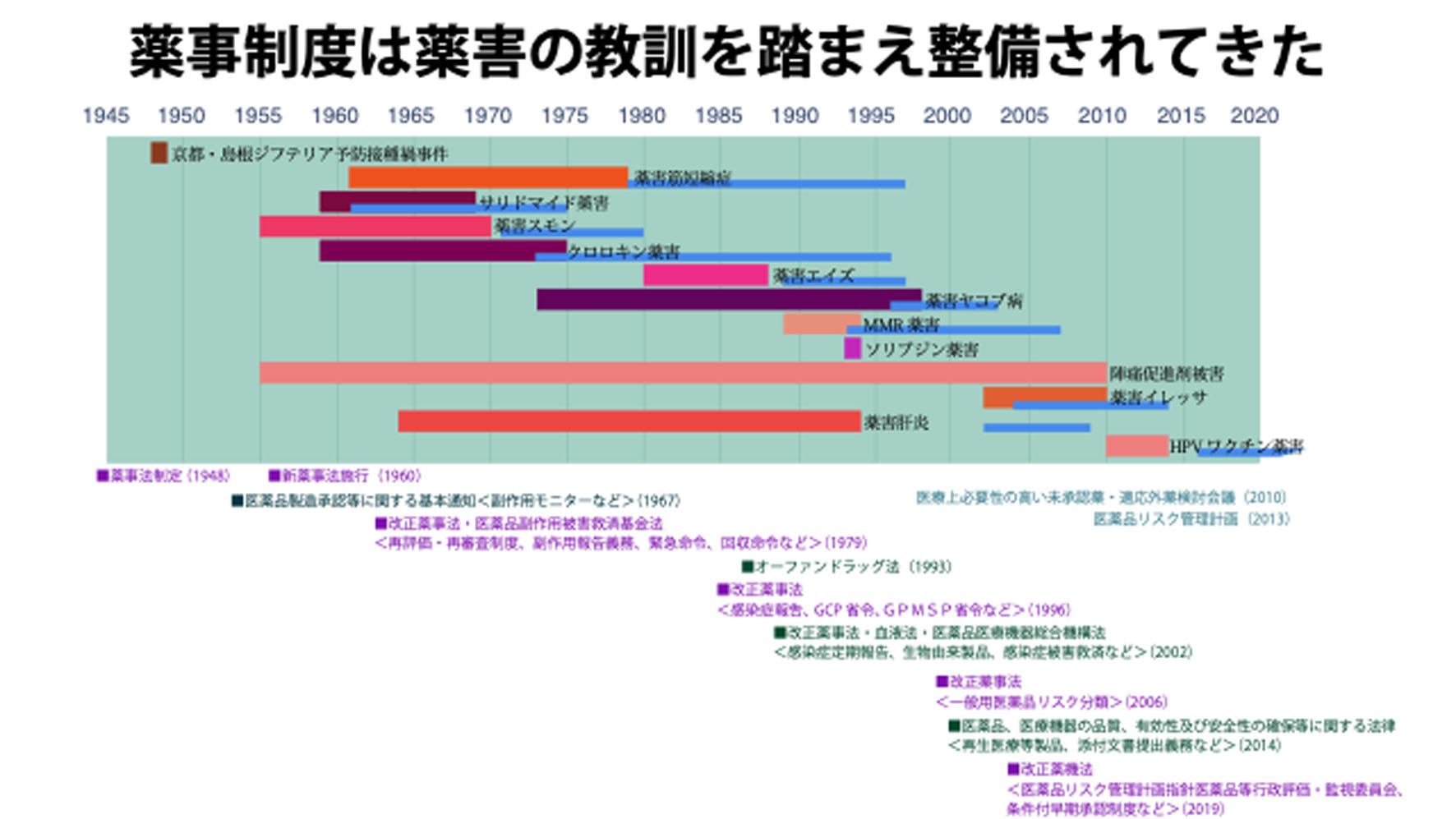
このスライドは薬事制度と薬害の関係です。私たち薬被連は、薬害被害を二度と起こして欲しくないと思いから、薬害被害の教訓を活かしたいとこれまで申し上げて来ました。実は世界的にも国内行政機関においても、教訓が活かされています。上の方の赤い線が被害や被害者が増えている期間です。青色の線は、訴訟がされている期間です。1960年代から今に至るまで継続して被害が起こっていることが分かると思います。一方、下に示したのは薬害被害に関連した薬事制度の変遷です。特にサリドマイド被害は世界的なインパクトがありました。下にあります日本で言えば1967年の通知です。世界で医薬品は有効性と安全性が大事なのだという今の常識、これが正にサリドマイド被害によって世界が自覚したということになります。この1967年が今の薬事行政の原点といえると思います。さらにスモン被害を踏まえて、その後の薬事法改正、1979年改正ですが、これこそが今でも常識である副作用報告義務とか、緊急命令回収命令といった今の建て付け、世界的にも1960年代から70年代にかけて今の常識的な枠組みが形成されたといえます。
その後、薬害エイズや薬害ヤコブが起きてきますが、そこで生物由来製品という概念ができるわけです。医薬品の原料の上流で、病原体があるかないかという規制が必要だと、つまり血液製剤で言えば、献血血液のルックバック、遡及調査システムが2002年の血液法に条文化されていくわけです。
その後も薬害が起こるたびにどこが悪かったのかということになっているというのがこの図であって、薬害被害は薬事制度と密接に関連して来たといえます。ただし、特に2000年代、右下の緑の2014年改正の議論のあたりから、医薬品を安全にしようと思ったら、臨床試験の数を増やして、サブグループを山ほど作って、時間をかけて安全性を調べればいいわけです。けれどもどんどん増やして行くとなると、コストも時間もかかるわけです。
この頃からドラッグラグという問題が生じてきます。すなわち欧米で使える医薬品が日本で使えないという、これを何とか改善するという問題がでてきます。つまり、より良い薬、より安全な薬をより速くということには誰も賛同しますが、意外にこれはコンフリクトするのです。だから急いで医薬品を承認すると安全性はどうかと。時間をかけて承認したら、逆にその医薬品を待っている患者さんが疾病によって死亡して元も子もなくなります。このバランスが非常に難しいということになります。
日本においては2000年代にかかってから、ドラッグラグの解消、特にはPMDAの組織の強化による審査機関の向上であったり、それから2014年改正においては再生医療等製品という仮免許制度、つまり仮承認制度を導入したり、期限付きの早期承認制度、2019年改正では、先駆け審査指定制度(先駆的医薬品等指定制度)と医薬品条件付早期承認制度という、それまで運用で行なっていた制度を法制化してドラッグラグを解消して、より良い薬を早く承認するという制度を整備してきました。開発メーカーにとっても、制度化によって計画性が立つということで歓迎されている制度であります。ただし、ことあるごとに私たちは「だからといって安全性を損なってはいけない」ということを繰り返し申し上げてきました。
ここからのスライドは、今回の緊急承認制度というものについて中心に意見を述べたいと思います。

これは実は患者さんが待っている薬を早く届けようという早期承認制度と筋の違う話であるということです。特例承認制度は1996年改正で導入された制度ですが、いわば緊急時に欧米で実績のある医薬品を緊急時に承認できるようにしようと言うのが、一つの理由付けで導入されました。これと似たような制度のように思えるのが今回の緊急承認制度ですが、思想的には特例承認制度の延長線上にあると解すことは問題ないと思います。問題なのは、緊急承認制度は国内開発の新薬等にも適用されるわけですから、そうすると医薬品承認における有効性・安全性評価基準が本質的に緩和してしまうという問題が生じるというのが大きな問題です。
先ほどの赤池参考人の方からもありましたし、同じ検討会に私も同席しておりましたので、この辺のことは理解するところですが、ただどの程度緩和すべきかというところで、有効性の推定と確認という言葉を使い分けていますが、推定と確認という言葉上、異なっていますが実際の運用になるとどこまで行けば推定されたとするのか、無限のグラデーションの中で決めていかなければいけないという問題があります。やはり、このような制度を作ることによって、いわゆるセキュリティーホール的なものになっては困るというのが私どもの主張です。

実は薬機法という法律自体がこういう緊急承認制度になじむのかという問題があって、私も検討会でしつこく主張してきたことであって、これは基本的には薬機法は製造販売業者に対する規制法ですから規制行政なのです。元々製販業者が医薬品として上市したいという動機付けからスタートして、医薬品を承認するために必要なデータを出すことになって、RCT(ランダム化比較試験)とか第三相試験とか、その中身に対して国がデータを評価審査し、足りない場合はこれを追加しろとか、承認はするけど市販後承認条件でこういうところにしか売ってはいけないとか、それから添付文書はちゃんと書きなさいとか、リスク管理計画RMPの中身をちゃんとしましょうということで、注文をつけて国が医薬品として承認するという流れです。
ところが今回の緊急承認制度は、国の方から医薬品として製薬企業に導入をお願いする局面があって、現にコロナ・ワクチンは優先的に売ってほしいとか、日本に輸出販売してほしいとお願いしたわけですから、これは全く出発点が異なっているので、本来薬機規律ではクリアできない問題があるわけです。
国が導入をお願いするような局面において、医薬品承認の主導権が入れ替わるかもしれない。売ってあげてもいいけど、あんまりうるさいことは言われたら困るとか、色々条件付けられたら、他で欲しい国がたくさんあるから日本に売らないとか、そういうことになることを懸念しています。
つまり薬機規律が機能しなくなるわけです。あくまで薬機規律は製販業者発で売りたい商品を承認するという建て付けなので、国が欲しい医薬品を、世界のどこかで手に入れてきて、医療現場へ持ってきたいから早く承認するということにはなじまないようになっています。なので原則を言えば、緊急承認制度という「承認」ではなく、「暫定使用許可」とするのが筋であると考えます。論理としてはそうしかありえないと繰り返し私は主張しております。アメリカではEUA(Emergency Use Authorization)の建て付けがありますが、これと比較しても日本型EUAとするには難しい問題があります。

検討会でも結局いろいろ意見はありましたが、取りまとめに最終的には賛成したわけです。その理由はスライドに細かいいろいろな法律が書いていますが、現行の医療関連法と統制技術的整合性という問題が常に日本の医療には生じてしまうという問題があります。
例えば承認した医薬品が全部薬価収載されて保険でカバーされるということが、日本の特徴なわけであって、医療システムと薬事承認がリンクしているわけです。そういうことから考えて、もし医薬品を適用外で使うという用い方を緊急承認ではなくて、緊急使用許可とした場合、ほかの法律をどう扱うかという問題が常に生じてしまうので、統制技術的整合性の観点からは今回のように薬機規律を利用するという考え方にも一定の理解を示すということが言えるかなと思います。それが故に検討会でも最終的には賛同させていただいたわけです。
特に保険療養のシステムは日本固有の皆保険制度で成り立っていますから、これ自体も相当細かい制度になっているので、皆保険制度関連とか、臨床研究法のようにオフラベルが特定臨床研究になるとか、薬機制度と関連して医療ヘルスケアシステムを統制する法律が並んでいるので、どのように整合させるかという話なので、逆に言えばテロ対策とか、コロナ・パンデミックに対する医薬品の使用というのは、本来は国家・政府レベルの統制システムの設計の問題であって、薬機法の中身だけで議論する問題ではないということをここで留保していただきたいと思います。

以上の懸念を踏まえまして緊急制度を薬機法改正に盛り込むにあたっては、お願いもしくは意見ですが、一つ目は緊急承認品目の承認はできるだけ速やかに対象群を設定したプロトコルによって安全性評価を求めます。ただしRCTを厳密にすると時間がかかるので、例えば5年後に安全性が分かりましたというのも困るので、ここはルールを決めて割と早期に有効性の評価をしてほしいと思います。それから安全対策については、ワクチンの場合、何千万人は難しいかもしれませんが、全数登録を原則としつつ、対象群から検出したシグナル(副作用など)を評価できる体制整備をお願いしたい。これ実は前にも議論をしておりまして、コロナ・ワクチンに関して結局、対象群つまり住民台帳を対象群とすればワクチンの安全性評価を日本でもできたわけです。しかしながら実はそれが出来ていないし、現状それをお願いしてはいますが、やはり市町村行政との関係になって医薬当局がいろいろ言っても協力してもらうのは難しいし、AMEDでという研究レベルじゃないでしょうというのが私の主張です。国家的に緊急事態に対応するものなのだから、医薬品の安全について国家的に保証するために、国家が持つ制度を動員してシグナル評価を出来るようにすることが政府の責任だろうと繰り返し主張しております。この場においても一番強く主張していきたいと思います。

リアルワールドデータという言葉も出ていますが、現にあるデータを使わないというのは非常に問題があると思います。そこの辺のところは、ぜひ議員の先生方にご理解頂きたいということです。それから緊急承認の場合、製薬企業と優先供給契約がされるわけです。その際に安全対策でいろいろ注文つけられるといろいろコストがあるから勘弁してくれっていうことを企業が言ってくる可能性があります。それを聞いて政府がPMDAに配慮するようになんて言う話は、薬機統制がゆがむのでやめていただきたいと思います。それから緊急承認品目の医薬品であることを国民がわかっていなければいけませんので、情報提供、できれば緊急承認品目という表示があるべきです。そしてインフォームドコンセントはやはり書面によってこれは普通の医薬品と違いますよと言うことを患者に徹底していただきたいと思います。

次に救済制度を導入することです。日本には世界に誇る医薬品副作用被害救済制度がありますし、予防接種法に基づく救済制度もあります。けれども医薬品との因果関係を完全に否定できない症例は救済されないとセーフティネットとなりませんので、この運用を徹底していただきたい。ただしこれには疑念がありますが、HPVワクチン、あるいはコロナ・ワクチンの被害救済においては本来救済されるべき国民が政策的意図によって除外されるようなことがあってはならないと思います。救済なのでメーカーからの拠出もあると思いますが、基本的には因果関係を完全に否定出来ない場合は、これはやめていただきたい。

最後になりますが臨床研究法という関連法で医師主導治験以外の医薬品開発においてもPMDAが対応する範囲を拡大することは議論されて、それはいいことだと思っています。国際的にはFDAが一括してやっていることは、臨床研究も治験も同じなわけです。それは理想なので今後もそうしていただきたい。また日本版のNIHを構想したAMEDは、今は単なるファンディングエージェンシーになっています。それがゆえに研究支援機能や評価機能が不足しています。それからHTA機関が存在していなくて、国立保健医療科学院の一セクションであるC2H(保健医療経済評価研究センター)が費用対効果を評価しています。 イギリスのNICEのようなものが無いということであって、これらの機関が充分に機能するような体制が必要だと思います。これらの機関が過去の薬害の歴史をちゃんと理解してほしいと思います。単に被害者だから言っているのではなくて、薬害を学ぶと今の医薬品の安全性向上に色々示唆するものがあると思っています。ぜひ歴史を踏まえた上で、これはレギュラトリーサイエンスでもありますし、日本の医療・国民の健康に寄与することを切望しております。以上です。ありがとうございました。